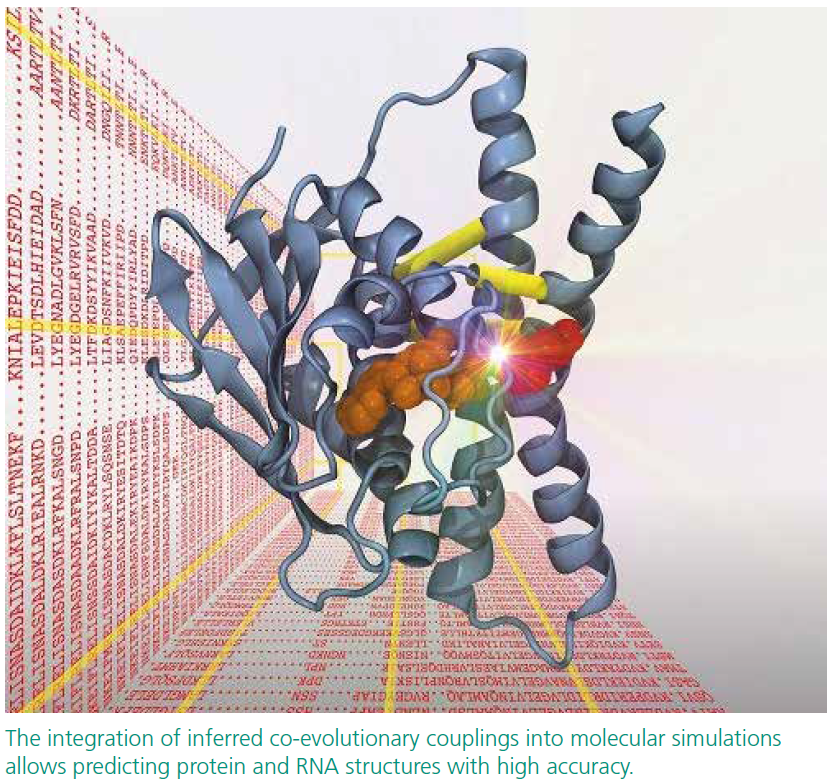
Protein Structure Prediction by Tracing Amino Acid Co-Evolution via Graph Theory
On the molecular level, proteins and RNAs are the Swiss army knives of life. They underlie all basic functions such as oxygen transport in the blood stream, providing stability to a spider’s web, muscle activity or enzymatic function in cells. Typically, their function is closely tied to their 3d structure. Yet the 3D structural characterization of many importan biomolecules is experimentally challenging. At the same time, genomic databases containing raw sequential ("1D") data about biomolecules grow exponentially. So why not exploit such databases? Over a few mugs of coffee, Dr. Alexander Schug and Jun.-Prof. Henning Meyerhenke
put their heads together. Combining their complementary expertise in biophysics and computer science, they developed a algorithmic framework for mining these vast amounts of raw sequence data with the goal of predicting biomolecular structures based on residue and contact co-evolution that are experimentally poorly accessible.
Alexander Schug has worked with a powerful statistical methodology called Direct Coupling Analysis, which can analyze evolutionarily closely related sequences to accurately predict pairs of residues in spatial contact within biomolecules. This information can guide the prediction of biomolecular structures when used in combination with molecular simulation. These methods are currently revolutionizing the field of molecular structure prediction, yet the molecular simulations remain computationally very expensive.
Henning Meyerhenke, in turn, is working on graph algorithms usually applied to analyze large complex networks. Two examples of such networks would be the friendship graph of a social network or the link structure of the world wide web. NetworKit, a highly performant opensource tool suite for scalable network analysis driven by Henning’s group, sounded well suited to process distance networks, which are similar to the data provided by co-evolutionary analysis, into biomolecular 3D models.
As a first step, Henning and Alexander managed to reproduce biomolecular 3d models in NetworKit in few seconds based on their distance networks. This is computationally considerably less expensive than biomolecular simulations, which are often running for days. In parallel to this work, they composed a long list of candidates for co-evolutionary analysis and structure prediction. Currently, both laboratories work on efficiently integrating the noisy co-evolutionary data into NetworKit and are also thinking about applying it to NMR data.